The NMR Facility at the Department of Chemistry and Biochemistry also supports macromolecular NMR research led by Prof. Rick Dahlquist and involving several other groups from Chemistry and MCDB, including the groups of Norbert Reich, John Lew, David Low, and Herbert Waite. The following illustrates some of the techniques in protein NMR research using examples mostly from collaborative work with researchers at the department.
Multi-channel, multi-nuclear pulsing capability is required in most biomolecular NMR research where the macromolecule is often isotopically labeled and heteronuclear NMR technique is applied. One key purpose of isotope labeling is to enhance the dispersion of the 1H signals by encoding the 1H signals with the through-bond correlated 15N or 13C chemical shifts. Without isotope editing, thousands of 1H signals from a macromolecule would be overlapping in a narrow 1H frequency range and detailed study is not possible. Equally true, 15N and 13C shift crowdedness is also reduced through correlation to adjacent 1H.
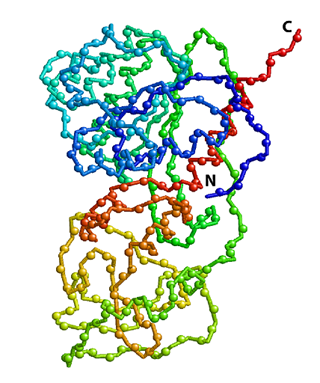 Crystal structure of HhaI, a 37 kDa DNA-binding methyltransferase. Positions of backbone amide 15N atoms are shown as spheres. The protein polypeptide chain is color coded to change from blue (N-terminus) to red (C-terminus). 15N-edited NMR spectroscopy, such as 2D 1H-15N HSQC shown in the right panel, allows the detection of all 15N-attached protons while all other proton signals are suppressed. Each sphere in the structure above represents a 1H-15N pair that gives a peak in the spectrum. |
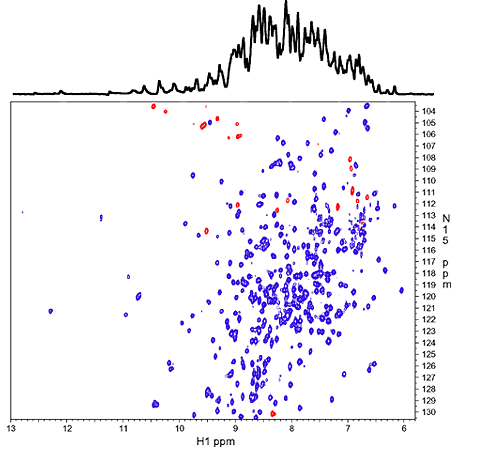 1D 1H spectrum of the amide NH region (top) and 2D 1H-15N correlation spectrum (HSQC). The overlapping 1H signals from over 300 NH protons in the 1D spectrum are mostly resolved along the 15N dimension in the 2D spectrum. Red peaks are NH resonances that are folded/aliased from outside the spectral window along 15N dimension. |
Isotope labeling of a protein is achieved when the protein is overexpressed in cells grown in isotope enriched minimal media. Typical labeling involves replacing naturally abundant 14N with 15N, 12C with 13C, and sometimes 1H with 2H (or D). In one type of 3D or 4D experiment conducted with an isotopically 15N/13C/2H triply labeled sample, the protein backbone amide 1H-15N nuclei are through-bond correlated with sidechain 13C nuclei while sidechain 2H nuclei are decoupled. Both 3D and 4D NMR data, once processed into the frequency domain, are simply 2D planes similar to any other 2D NMR spectrum, except that there are many of these 2D planes and each is indexed by a 15N or 13C chemical shift in a 3D spectrum, or double indexed by both 15N and 13C shifts in a 4D spectrum.
Another important advantage of the isotope editing technique is fundamental to structural biology. 1H signals alone are not sufficient to address the wide range of questions related to the structure and biological function of the molecules. A number of important NMR techniques involve studies using the isotopes directly. These include, among others: (1) secondary structure prediction based on 13C chemical shifts in a folded protein, (2) protein dynamics through measurement of 15N or 13C relaxation properties, and (3) structure confirmation and modeling with residual dipolar couplings of 1H-13C and 1H-15N.
It is well known that when a protein folds into a specific 3D structure from
a random coil polypeptide, chemical shift dispersion of both 1H and
13C nuclei increases, reflecting more diverse local chemical
environments of the nuclei and specific atomic contacts. 1H and
13C chemical shifts are sensitive to local secondary structure
formation such as helix, strand or random coil. Chemical shifts of backbone
13CO and 13C of sidechain CαH and CβH for
random coil peptides have been measured and compared with chemical shifts from
residues in well-formed secondary structures in proteins. It has been well established that these
chemical shifts correlate well with the type of secondary structure. These
findings lead to the popular secondary structure analysis technique named
chemical shift indexing or secondary shift
analysis.
The secondary chemical shifts are first calculated as the
differences between the measured values from the protein and the random coil
values. These differences are plotted as a function of residue number.
Consecutive positive or negative deviations are classified as either helix or
strand structure with the degree of deviation corresponding to a level of confidence about
the structural assignment. Such an approach has a remarkable accuracy, particularly when
coupled with a database search of short peptide sequence against a set of PDB
structures, as implemented in the program TALOS. Proton secondary shifts of
CαH groups also correlate with protein secondary structure. However,
13C chemical shifts are more reliable in this analysis due to its
wider chemical shift ranges and more types of 13C nuclei, including
Cα, Cβ and CO, available for this analysts.
 Structure of a flagellar motor protein FliM |
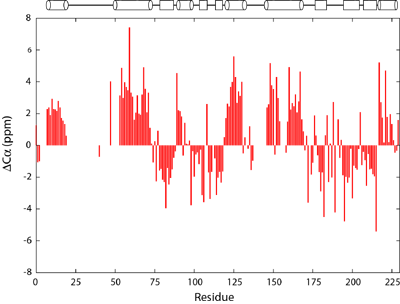 Deviation of Cα chemical shifts from average values from random coiled peptides. The secondary structure of FliM is schematically drawn above the figure with helices denoted by cylinders and strands by rectangles. |
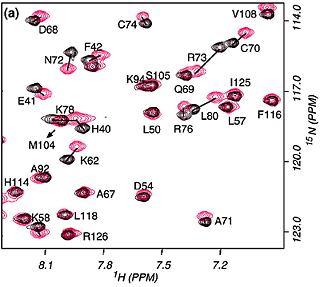
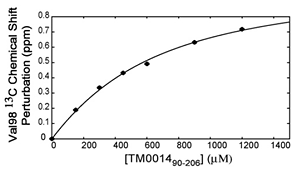
When a protein interacts with a small molecule ligand or another macromolecule, perturbations to the NMR crosspeaks of the interface residues are often observed. These changes are used to identify the binding interface. This method, called "chemical shift perturbation mapping", is widely used under the assumption that the shift perturbations, due to either structural changes or close contact, are localized in the macromolecule. While this assumption is proven true in most cases, large-scale chemical shift perturbations could obscure the actual binding site, rendering the data analysis less reliable. This situation could occur due to global structural changes or multiple binding sites, etc.
The binding assay is often done using heteronuclear NMR with one partner isotope labeled to observe its signals and the other unlabeled or deuterated.
As in all binding studies under NMR condition, the scale of exchange rate (Kex) between the free and bound forms relative to their chemical shift difference (ΔCS) has a strong influence on how the titration data appear and should be interpreted.
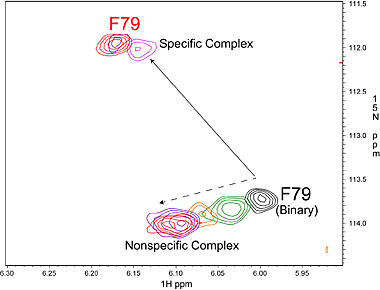
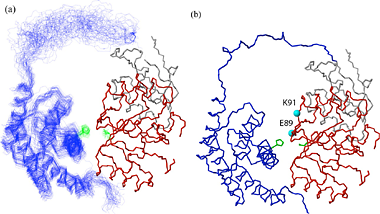
Specific (strong, slow-exchange) and non specific (weak, fast
exchange) interactions between HhaI and DNA. When a piece of
non-cognate DNA resembling the cognate DNA in sequence is added to
HhaI, the protein resonances display both specific and nonspecific
binding characteristics. Shown on the right is a superposition of a set
titration spectra of HhaI showing Phe79 going through a transition from
the free form (binary) to the DNA-bound specific complex and
non-specific complex, ending with a mixed population of specific and
nonspecific complexes. The continuous movement of the nonspecific
complex peaks along a straight line is typical of weak binding with
fast exchange rate. In the slow exchange case, the intermediate peaks
are not observed and the bound peak simply gets stronger as the free
protein peak gets weaker until binding is saturated with DNA.
|
Keep in mind that the exchange rate and binding affinity may fall anywhere in between these three cases. In the fast-exchange case, the kD of the binding can often be obtained from fitting the chemical shift as a function of substrate concentration. In the borderline cases, however, detailed analysis may not be always possible.
|
|
|||||||||||
Solution structure determination is one of the main applications of NMR techniques. It not only compliments the X-ray structure but importantly it also provides a more native-like structure under solution state. In some cases, the protein fails to crystallize and NMR technique becomes the only tool to provide a high-resolution structure.
The structure determination process typically starts with overexpression of isotope-labeled proteins, followed by a series of 3D and/or 4D NMR experiments that enable 1H, 15N and 13C resonance assignments to specific nuclei in the protein. The sequential assignment process is done on the computer by examining sequentially correlated crosspeaks to establish their sequential relationship. Once the sequential assignments are made, NOESY (Nuclear Overhauser Effect Spectroscopy) data are analyzed and assigned to proton pairs.
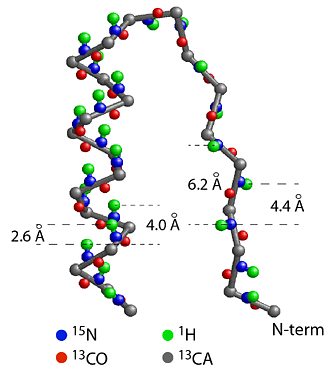
(a) Section of CheY structure formed by a strand, a turn and a helix starting from the N-terminal end. The helix is a more compact structure and the strand is an extended structure, causing the distances between the amide protons (green) to be short between neighbors in a helix and much longer in a strand. This leads to strong amide-amide proton NOEs for a helical structure and weak NOEs for a strand structure. The patterns of amide-amide and amide-CαH proton NOEs are good indicators of secondary structure. 1/r6 rule: NOE intensity drops off as a function of 1/r6 where r is the distance between the protons. The rapid dropoff only allows NOE between protons of < 5 angstrom, making NOE a sensitive measure of short distances. |
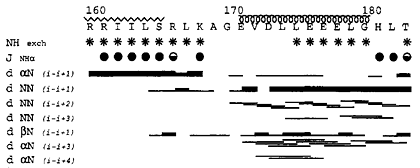 (b) Summary of hydrogen exchange experiment, NH-CαH 3JNHα coupling constants and NOE data. In a hydrogen exchange (HX) experiment, the protein sample in H2O is rapidly switched into a D2O buffer and an 1H-15N correlation spectrum is collected immediately. The peaks from the amides protected from HX, mostly due to hydrogen bonds and/or burial, remain in the spectrum long after (mins, hours, or days) the exchange while those who disappear quickly are presumed not hydrogen-bonded or buried, therefore have been replaced with deuterons and are not observable. NH-CαH 3JNHα coupling constants correlate well with the dihedral angle Φ according the Karplus equation. Large (> 8 Hz) J-values, shown as filled circles, indicate extended strand structure; smaller (< 7Hz) values indicate helical structure. The NOEs among various neighboring protons (NH-NH, CβH-NH, CαH-NH) are shown by black lines. The thickness of the lines corresponds to strength of the NOE. The secondary structure assignment based on these data are drawn above the figure. |
NOE occurs through the dipole-dipole interaction between protons within a short distance, typically within 5 Angstrom. These interactions are presented in the NOESY spectrum as crosspeaks linking two protons. Distance information is obtained from NOESY spectra and is applied in the form of distance restraints in a structure calculation program such as X-PLOR to fold the polypeptide chain into a 3D structure. Other constraints in the calculation include H-bonds related to secondary structure, determined by a combination of chemical shift index (see above), dihedral angle measurements, NOE patterns, D2O hydrogen exchange experiments, etc.
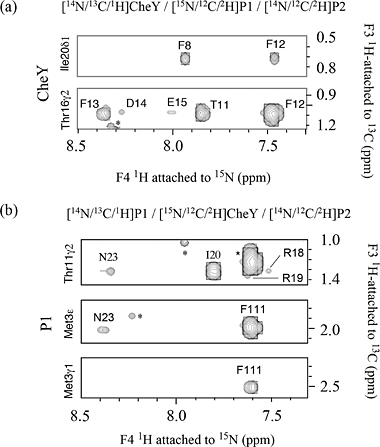
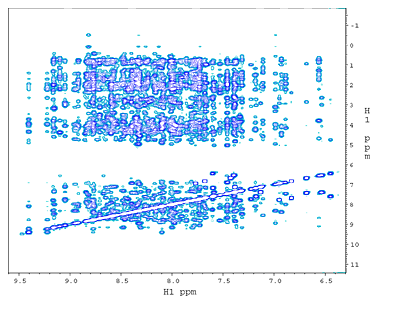 (a) 2D 15N-edited NOESY from a ~130 amino acid, 15N,13C-labeled phosphotransfer domain (P1) of chemotaxis histidine kinase CheA. The NOE crosspeaks here are between 15NH amide protons and all other protons. |
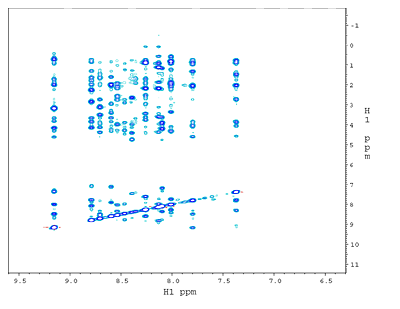 (b) 1H-1H NOESY plane at 120ppm along 15N from a 3D 15N-edited NOESY spectrum. The dramatically reduced crowdedness of the 1H peaks allows the assignments of thousands of NOE crosspeaks to individual proton pairs. The 2D crosspeaks in (a) are spread over 64 1H-1H planes each with a unique 15N shift . |
||
 (c) Slice at apparent shifts of 15N: 120ppm and 13C: 62.5ppm from 4D 15N,13C-edited NOESY. All peaks seen are NOE crosspeaks between 15N-attached protons and sidechain 13C-attached protons. Compare this figure with the 1H-1H plane in the 3D 15N-edited NOESY in (b) from the same 15N slice. Note the reduction in the number of peaks in the 4D spectrum due to the additional 13C editing, as shown by the appearance of the boxed peaks but not others among the vertical strips of peaks having NOE to these two NH protons. |
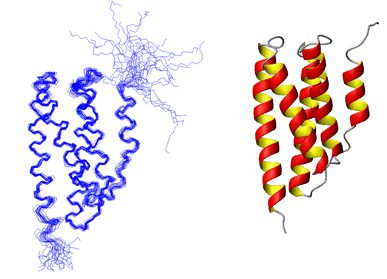 (d) P1 Structure determined using 2759 NOE distance constraints along with some H-bond and dihedral angle constraints. On the left is a best-fit superposition of 25 structures. On the right is a ribbon diagram of the average helix bundle structure. |
||
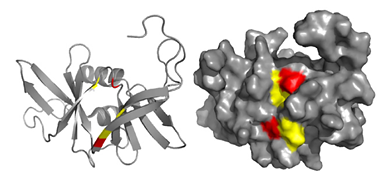
Structure of P1-CheY Complex
|
|||
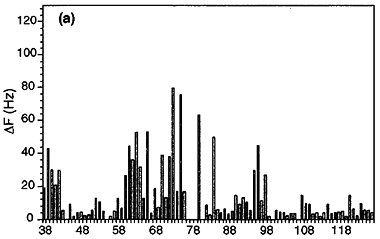
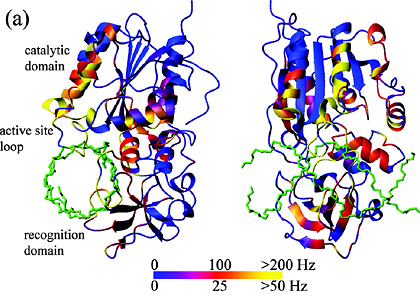
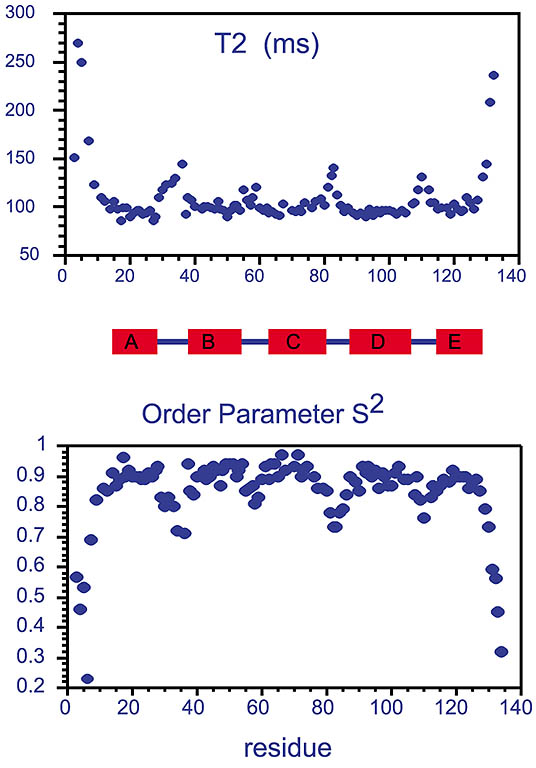 Measured T2 values and extracted order parameters from the phosphotransfer domain of the histidine kinase CheA.
Increased T2 values, often found in the terminal ends and in turns between the rigid helices (shown as A-E rectangles),
reflect more flexibility of the protein backbone in these regions. This trend is also reflected in the extracted order parameter based
on Lipari-Szabo's model-free approach assuming the relaxation of the amide 15N spin is caused by
dipolar coupling from the amide 1H in addition to chemical shift anisotropy, both modulated
by molecular tumbling and internal local dynamics.
|
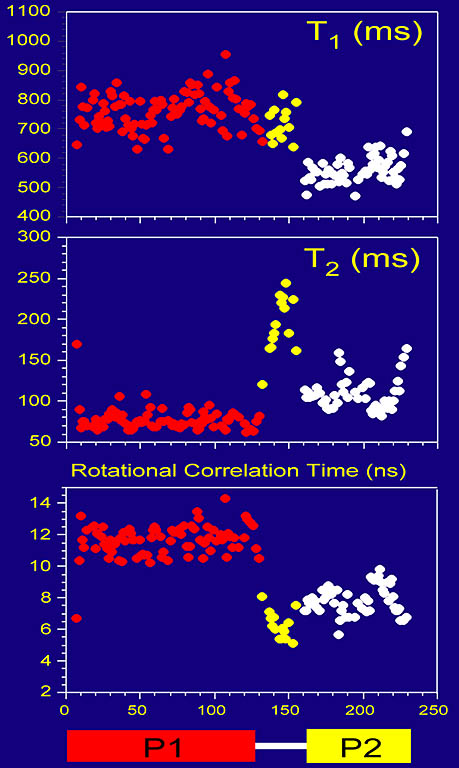
Protein backbone dynamics of a 233-residue fragment of chemotaxis histidine kinase CheA revealed
by measured T1, T2 and extracted overall rotational correlation times of backbone amide 15N spins. The two
domains of the fragment are joined by a flexible linker with little contact between the domains, evidenced
by different dynamics parameters and high mobility in the linker. The N-terminal domain (residues 1-134) is twice as large as
the C-terminal domain.
|
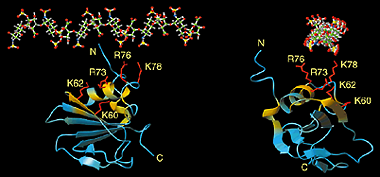
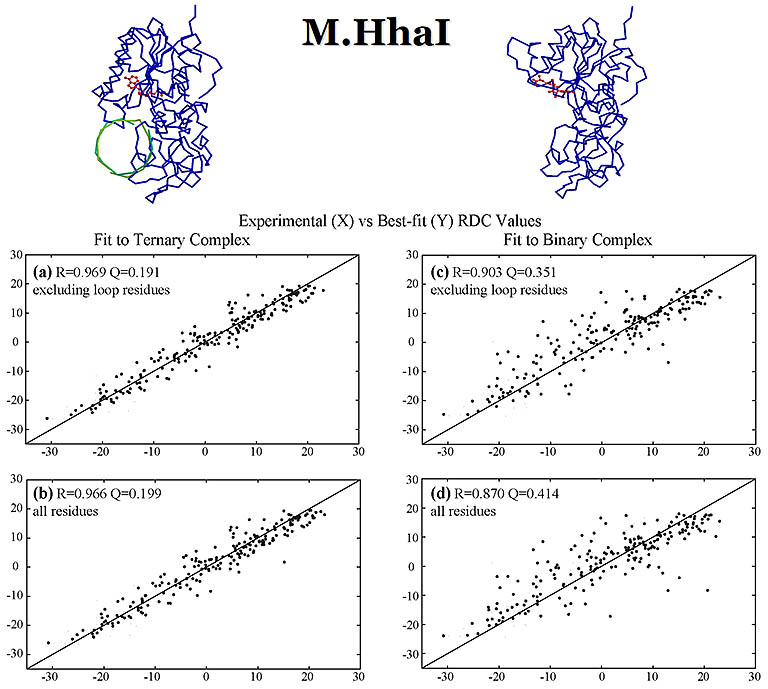 RDC values (Hz) were measured for DNA methyl transferase M.HhaI bound with a DNA fragment. These values were fit with the crystal structures of
the apo- and DNA-bound structures, both bound with a cofactor. The measured RDCs are consistent with the DNA-bound ternary crystal
structure and confirm a large closure movement by a 20 amino acid active-site loop to lock down the DNA as observed in the crystal structure.
The DNA fragment, looked down towards its helix axis, is shown in green. The cofactor is in red. The alignment media
used was a mixture of alkyl-polyethylene glycol C12E5 and hexanol.
|
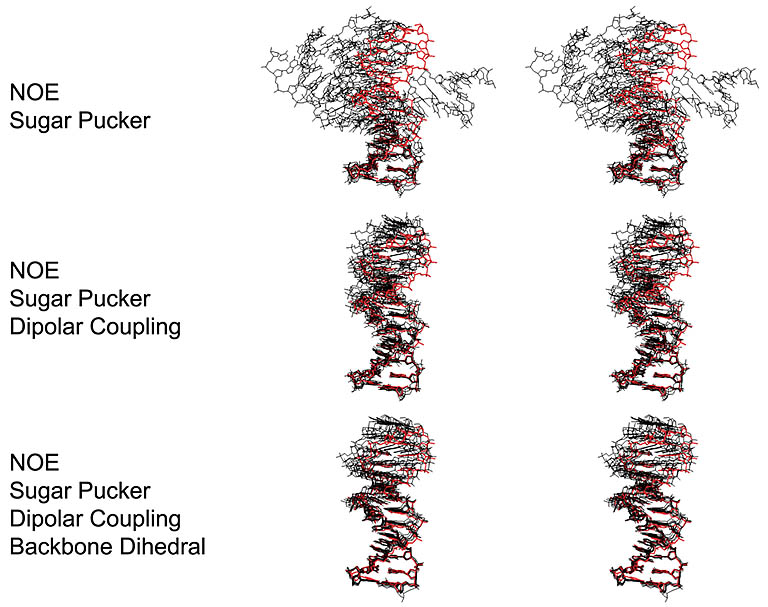
Structure refinement using RDC values as constraints. The precision of DNA structure improves
with the inclusion of RDCs in addition to other constraints.
|
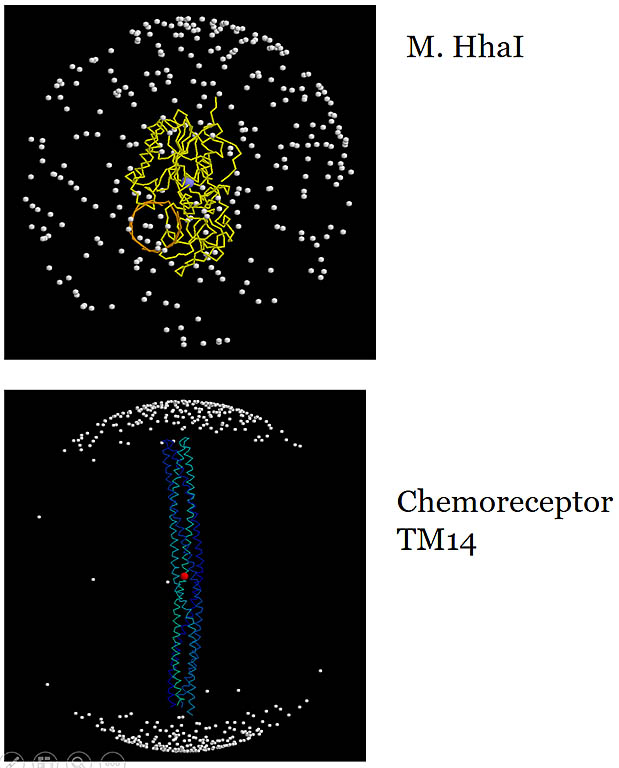 Amide 1H-15N bond vectors in the molecular frame.
The vector from the center of the sphere to each dot is an amide 1H-15N vector. The useful information
content in measured RDCs relies on a wide, more uniform distribution of the vector direction reflecting the molecular shape. The non-spherical shape
of the molecule enhances asymmetrical molecular tumbling and enlarges measured RDCs. Meausred RDCs in the
TM14 chemoreceptor fragment would be heavily biased along one long axis and don't provide independent data content addressing
the other perpendicular axes.
|
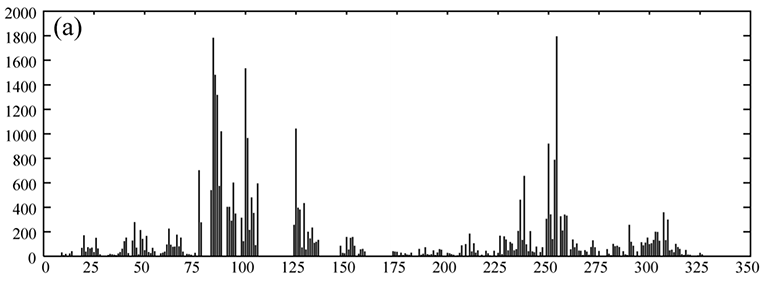
 Amide 1H-15N bond RDC measurement for the phosphotransfer domain (~ 134 aa) of the histidine kinase CheA. The one-bond 1J + RDC value (~ 90 - 100 Hz)
shown along the vertical 15N dimension is measured in two
separate experiments to give the two split peaks, overlayed in black and red. The difference of this value in the presence
and absence
of the alignment media of Pf1 phage particles is taken as RDC.
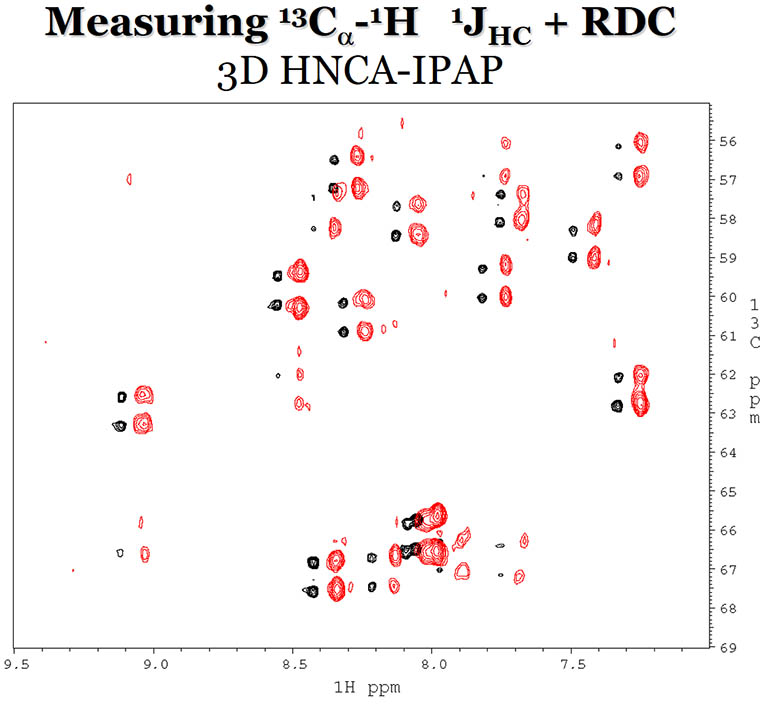 RDCs between 1H and 13Cα are
measured in a 3D HNCA experiment with (red) and without (black) Pf1 phage as the alignement media.
|
TROSY is widely used for large molecules when they are deuterated. Deuteration removes other 1H dipolar couplings that contribute a background broadening to the peaks. Generally, deuteration (or 2H labeling) is necessary in cases where the large size of the macromolecule leads to shortened T2 relaxation time due to strong 1H dipolar couplings and slow molecular tumbling, consequently lowering signal-to-noise ratio. 2H (or D) has only a very small magnetic moment therefore a small dipolar interaction with other nuclei. Replacing some 1H with 2H in a protein often dramatically increases T2 of 1H and 13C nuclei of interest and enhances the sensitivity of most experiments. In one situation the uniform deuteration occurs for all protons except the backbone amide NH and other labile protons to enable their detection. In another scheme, using special protein production techniques, only the methyl CH3 groups are 13C-labeled and protonated, leaving all other carbons unlabeled and all other protons deuterated except the labile protons. These labeling schemes enable NMR experiments on macromolecules or complexes of 30 kDa or larger.
The advances in NMR probe technology, increase in field strength, improving isotope-labeling techniques and the development of TROSY have together enormously broadened the biomolecular NMR field in the last two decades. The size of protein or complexes that can be studied by NMR has increased to several hundred kDa. The precision of NMR structure determination has reached level comparable to X-ray crystal structure in many cases.
Hongjun Zhou, updated 2019