Chemical Shift Referencing
Background
Chemical shift (use symbol δ) values are based on historical conventions. While they reflect physical, electronic
shielding (use symbol σ) of the nuclear spins,
their values carry only relative meanings at best. Note that IUPAC recommends against the use of the term "chemical shielding" in preference for
electronic or magnetic shielding. Both electronic shielding and the derived chemical shift are tensors, because
their values are directional in the molecular frame. In isotropic solution, however, fast and uniform molecular tumbling diminishes the directional dependence,
and reduces the dominant contribution of the tensor to an average, scalar value in the Hamiltonian. The residual components of the tensor (chemical shift anisotropy) contribute to nuclear spin relaxation. Here, we exclusively discuss scalar
chemical shifts (also called isotropic shifts) in solution state NMR.
When referencing chemical shift of any nucleus, including 1H, 19F and X-nuclei, we follow one of several methods recommended by
IUPAC whenever possible (proposed in 2001 and updated
in 2008).
IUPAC recommends the use of a "unified chemical shift scale" and that ALL
chemical shifts be referenced against the methyl 1H signal from very dilute TMS (<1%) in organic solvents
or DSS in aqueous solutions,
either directly or indirectly. The TMS reference is called the "primary reference". Imagine that all nuclei share
the same frequency scale on a spectrometer, i.e. ranging from 0 to 500 MHz on a 500 MHz NMR spectrometer.
In principle, only ONE referencing point is needed and is sufficient to denote all chemical shift variations for any nucleus.
This is the foundation of the "unified chemical shift scale" concept.
Initially, IUPAC envisioned replacing the use of the historical chemical shift with the ratio (Xi, Greek letter Ξ) of the resonance frequency of any nucleus relative to the methyl 1H frequency of
TMS. However, this reporting method
has never gained traction and has been mostly ignored across the field. Currently, the method where
each nucleus uses its own, historical 0 ppm reference compound remains in widespread use.
Traditional, individual nucleus 0 ppm reference is called "secondary reference" following the
introduction of the unified scale with the TMS 1H resonance as the primary reference. Most chemical shift referencing is now done under the umbrella
of the "unified chemical shift" scale but still uses the traditional reference
compound for each nucleus. From here on, all Xi (Ξ) symbols and
values only refer to those of the historical reference compounds. Note that alternative secondary references should be avoided
as much as possible (see note below), with the exception for 15N/14N.
To continue to use such historical referencing compounds and also to comply with other
IUPAC recommended conventions, IUPAC has compiled tables of the Ξ values of all NMR active nuclei
(i.e P31 in H3PO4, see Tables 1 and 2). The Ξ values
are also listed here in Varian instrument's nucleus table (2nd column).
This ratio (Ξ, often expressed as % of TMS 1H frequency), measured to high precision and
field independent, allows indirect establishment of the zero ppm point of the traditional (secondary) reference compound of any nucleus in the table
once the resonance frequency of the TMS 1H signal (primary) is known. Moreover, based on a similar approach, using the known lock (often 2H) frequency
or generally any other nucleus resonance with a well established chemical shift, the frequency of the reference signal of any other
nucleus in the Ξ table can be calculated in the absence of the secondary reference compound.
This method is universally applied on modern NMR spectrometers and in most processing programs.
IUPAC further recommended that these Xi (Ξ) values be frozen, immutable regardless of future more "accurate" measurements.
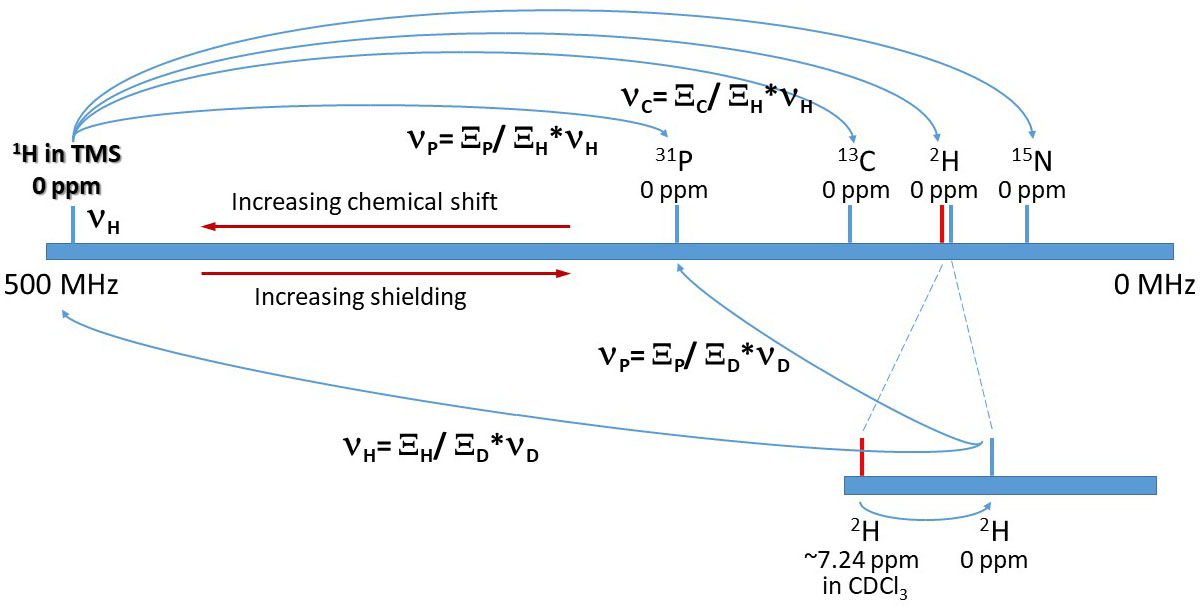
Chemical Shift Referencing on the Unified Scale
Here, ν
H is the precise frequency of the
TMS 1H resonance. ν
X is the precise frequency on the same instrument of X nucleus in its historical reference compound.
Ξ
H is set at 100 in the Ξ table. Ξ
X is given as a percentage of Ξ
H.
Top: With the Ξ value and the observed TMS 1H resonance, the 0ppm reference position of any other nucleus
in the Ξ table can be computed.
Bottom: In the absence of TMS 1H signal, if any other resonance with a known ppm value (here the 2H lock signal
from CDCl3 at ~ 7.24 ppm in red) is observed, the 0ppm position of any other nucleus can also be computed with the same Ξ table. Note the IUPAC convention that higher frequency
is to the left and corresponds to higher ppm and less shielding relative to the reference nucleus.
Chemical shift and electronic or magnetic shielding vary in opposite direction by definition (see pp. 77-78 in
IUPAC Further Recommendations).
Referencing using the pre-determined Xi (Ξ) ratios of the historical reference compounds
along with an observable signal of known ppm value dramatically simplifies
chemical shift referencing because there is no need for the
presence of the reference compound or resonance. This indirect referencing method (in the absence of the primary TMS signal) is
sufficient for most routine analytical NMR work, unless a strict comparison of shift values across multiple samples or conditions
is needed. In that case, extreme care has to be taken regardless of what reference method is used.
Recommended Referencing Method
For most routine NMR work when rough referencing is sufficient and TMS (or DSS) is not present in the sample:
- If a solvent signal (1H or 13C) in the spectrum can be located and has a known
ppm value in the solvent table, use this signal.
- If no residual solvent signal is available or can be reliably identified, use the lock signal (2H or D) to reference the
spectrum of any nucleus.
- Note that in most 3rd party software, including Mnova, and most manufacturer's software, the default (often automatic) referencing is through the 2H lock signal.
If a more precise and consistent chemical shift reference is desired:
- Add 1% or less TMS or ~ 1 mM DSS in aqueous solvents to your sample. Collect the 1H spectrum and reference it by
setting the TMS or DSS methyl peak to 0 ppm. To reference any X-nucleus spectrum from this sample, see below on how to do so
using the 1H spectrum in VNMRJ (Varian software) or in Mnova.
- DSS may be deuterated at all CH2 positions if those resonances interfere with data analysis. DSS-d6 is available
from Sigma or Cambridge Isotope Lab.
Other choices (NOT recommended in most situations unless TMS or DSS reacts or interacts stronlgly with other chemicals in the sample):
- (1) Substitution method: use a a separate NMR tube containing the reference compound and collect a separate reference spectrum. Any change in
locking, shimming and tuning between the two samples should be avoided.
- (2) External referencing: use a reference compound sealed in a capillary tube inside the same NMR tube.
External referencing with the reference compound separated from the sample of interest does not give better reference
accuracy, and is tricky to apply properly due to field correction factors, the required steps regarding locking, shimming, etc.
They should only be used in very limited situations. For more details and pros and cons of each referencing
method, see the IUPAC documents linked above.
Note that TMS has a melting point of -99C and a low boiling point of 27C. Outside
this temperature range, you could still use TMS signal detected within the above
temperature range, due to the low thermal sensitivity of TMS shift.
Alternatively, but less precise, try to use the lock signal from the solvent to
roughly reference your spectrum.
Description of Referencing Method in Publication
If a direct, internal or external reference compound is present and used, this should be stated. The description needs to be
clear about how the referencing is done and indicates any condition different from room temperature
and one atmosphere pressure. If an alternative secondary reference is used, i.e. NH3 is used for 15N instead of CH3NO2,
this should be stated clearly. In the following, TMS may be replaced with DSS in aqueous solutions.
- If TMS is present in the sample, and is used as the 0 ppm reference for 1H, simply state the 1H spectrum is referenced with
the TMS 1H resonance.
- If TMS is present in the sample, a 1H spectrum is collected and referenced with TMS, and is used to reference a X-nucleus spectrum (see below):
"The X-nucleus (i.e. 13C) spectrum is referenced with the internal TMS 1H resonance following IUPAC recommendations."
- If residual solvent signal (such as 1H of CDCl3 at ~7.26ppm) is used to reference the spectrum, the description should be
something like this:
"The residual 1H resonance from the deuterated solvent is used to reference
the 1H spectrum, according to IUPAC recommended method and the manufacturer's protocols (add literature references above if necessary)."
- If the 13C solvent signal is used to reference the 13C spectrum, the following describes the method:
"The 13C spectrum is referenced through the solvent 13C resonance (or with the solvent 13C resonance set to XXX ppm) according to IUPAC recommended
method and the manufacturer's protocols (add references above if necessary)."
- If a spectrum from any nucleus is referenced via the "By Solvent" method, the following describes the method:
"The spectrum is referenced through the solvent lock (2H) signal
according to IUPAC recommended method and the manufacturer's protocols (add references above if necessary)."
Chemical Shift Referencing in Mnova
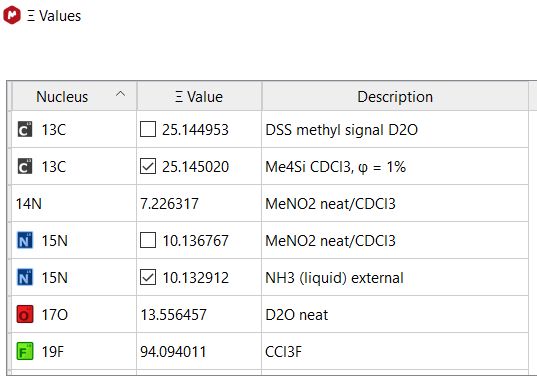
CAUTION: Do not modify the Xi values as suggested by Mnova that you may do so. Use IUPAC Xi (Ξ) table strictly.
For additional information, see Chapter VIII, pages 203-209, Mnova manual.
To reference the spectrum by the lock 2H signal (only if the sample is locked with the proper 2H solvent)
Go to Analysis→Reference→Reference By Solvent.
To reference an X-nucleus spectrum using a properly referenced 1H spectrum collected on the same sample and at the same field, ideally sequentially
without changes in lock and shims:
Load and process 1H and X-nucleus (including 19F) spectra. Reference 1H spectrum properly, ideally using an internal TMS signal as recommended.
Go to Analysis→Reference→Absolute Reference .... At the top, select the 1H spectrum to be used as the reference spectrum. Then, check the
spectra or dimensions to reference below and click OK.
For 15N, to change the secondary reference between CH3NO2 and liquid NH3, open the Xi (Ξ) values table under Analysis→Reference→Absolute Reference ....
Select the proper secondary reference for 15N, then re-reference the spectrum. A roughly +/-380.50 ppm change in 15N chemical shift
is expected after the switch.
| Mnova Reference Menu and Ξ table |
 |
 |
Varian Chemical Shift Referencing
Reference By Solvent (2H) Signal: (for all nuclei, including 1H, 19F, and X-nucleus):
Varian command: setref or click Process→Default→By Solvent
CAUTION: For some deuterated solvents, there are multiple deuteron peaks. This may cause reference errors with
the through-solvent method because the lock position may be one of several positions. In this case, for reliability, you can
transfer the reference of a properly referenced 1H spectrum to any other spectrum with the command mref.
mref(source_exp#,target_exp#)
To use a referenced 1H spectrum (ideally with an internal TMS signal) to reference any other X nucleus spectrum:
- Example: mref(1,2) reference the
spectrum in exp2 (i.e. 13C) using the 1H reference in exp1
reff1, reff2
- use referenced direct dimension (1H) to reference indirect dimensions in 2D or 3D.
Differences Using TMS or DSS as Primary Reference
The 1H shift difference between TMS and DSS is generally < 0.03 ppm in various solvents. On the unified scale, the X-nucleus reference frequency is:
νX0 = ΞX/ΞTMS*νTMS
Switching from TMS to DSS as the primary reference results in essentially the same small ppm change for the X nucleus.
Except noting the primary reference as DSS 1H signal (1H shifts denoted as δDSS), no other adjustments are
needed when DSS is used instead of TMS to reference other nuclei. IUPAC states:
"For most purposes, these differences are negligible (falling well below the anticipated range
of solvent effects), and data from the TMS and DSS scales may be validly compared without correction
for the different 1H reference."
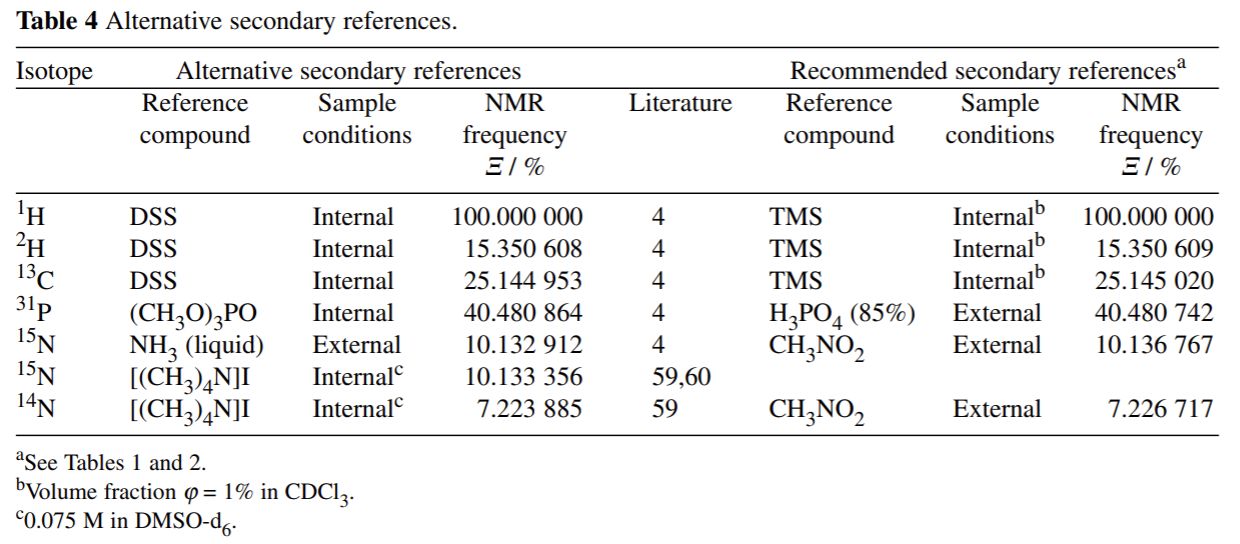
Alternative Secondary References
Alternative secondary references that differ from the standard IUPAC references should be avoided as much as possible. One
exception is 15N/14N where liquid NH3 is commonly used as reference instead of CH3NO2, especially for protein NMR and
studies in aqueous solutions. Which alternative secondary reference is used should be clearly stated. In the case 13C,
if DSS is used as the secondary reference (0ppm for 13C) instead of TMS, the 13C shift increases by ~ 2 ppm. For 15N, if NH3
is the reference instead of CH3NO2, the 15N shift increases by ~ 380.5 ppm.
IUPAC Alternative Seconadry References
 |
Immutability of Xi (Ξ) Values
IUPAC recommended against (and it makes sense) any changes to the published Xi (Ξ) values. This goes back to the
fact that chemical shifts are not intended to be measures of nuclear spin shielding and they have only relative
meanings.
From the updated recommendations in 2008:
IUPAC recommended that the values of Ξ in the 2001 document [4]
"are not subject to future change arising from remeasurement even where this results in increasing accuracy for the reference compound in question." This recommendation echoed a similar recommendation by IUPAC/IUBMB [3]. The immutability of these Ξ values has sometimes been questioned, since
it might appear that newer, more accurate, results should be incorporated, as is the case with most scientific data. However, the principal purpose for the tables is to provide a consistent set of numbers that
can be used to provide a link between data for various nuclides referred to the universal TMS reference
and results already in the literature where each nuclide is referenced separately. In general, the values
of Ξ that are given here are of sufficient accuracy relative to previously published data to permit valid
comparisons. To allow changes to be made from time to time in these values would result in inconsistent and confusing comparisons.
In other words, unless we can routinely, precisely measure vacuum (a naked nucleus), we should be satisfied with the consistent, relative measurement.
References
- NMR Nomenclature. Nuclear Spin Properties And Conventions For Chemical Shifts. R. K. harris et al (2001) Pure Appl. Chem., Vol. 73, No. 11, pp. 1795-1818.
- Further Conventions For NMR Shielding And Chemical Shifts. R. K. Harris et al (2008) Pure Appl. Chem., Vol. 80, No. 1, pp. 59-84.
Hongjun Zhou,
updated April 2022